Table of Contents
Anvi'o
Anvi'o is an advanced analysis and visualization platform for 'omics data.
- Anvi'o project page http://merenlab.org/software/anvio/ “Anvi'o in a nutshell”
- v5.1 “margaret” 소개 https://github.com/merenlab/anvio/releases
- 현재 bioconda로 설치되는 최신 버전은 v4.0이다. 이를 확인하려면 'anvi-profile –version'을 입력한다. — Haeyoung Jeong 2018/07/06 11:52
{kind=link}
Installation (bioconda)
Please note, anvi'o uses Python 3 exclusively. ⇐ 프로젝트 사이트에서 글을 먼저 보았어야 한다! Bioconda를 이용한 최초의 설치에서는 python 2.7을 기반으로 한 environment를 새로 만들어서 작업을 했었다. 테스트 스크립트의 실행에서 접했던 몇 가지의 문제가 python 버전 때문이었을까? 두번째 시도에서는 기존에 있던 py35 환경에 anvi'o를 설치하였다.
# source activate py35 (py35) # conda install -c bioconda -c conda-forge anvio diamond bwa # anvi-self-test --suite mini # anvi-profile --version Anvi'o version ...............................: unknown <= 왜 unknown이지?? Profile DB version ...........................: 23 Contigs DB version ...........................: 10 Pan DB version ...............................: 8 Genome data storage version ..................: 6 Auxiliary data storage version ...............: 2
anvi-self-test 실행을 통해서 설치 문제점 해결하기
- prodigal의 버전을 맞추어야 한다.
- [py27에서만] “ImportError: cannot import name wsgiserver”라는 에러가 나면 다음을 실행한다. 참고 사이트
pip install "cherrypy>=3.0.8,<9.0.0"
이상의 문제가 다 해결되었다면 anvi-self-test –suite mini를 실행한 뒤 웹 브라우저가 뜨면서 결과가 나타나야 한다. 그러나 크롬이 아니라면 결과가 제대로 표현되지 않는다(Installing anvi'o 페이지 가장 위에 있는 A NOTE ON CHROME 참고). 하지만 CentOS는 더 이상 크롬을 지원하지 않는다! 따라서 해결 방법은 크롬이 아니라 크로미움을 설치하는 것이다.
[CentOS 6.5] Chrome Browser (Chromium) 설치 ← 관리자가 아닌 일반 사용자 권한으로 크로미움 브라우저를 실행하도록 설정하는 방법도 같은 문서에 나온다.
anvi-self-test가 성공적으로 실행되면 시스템의 디폴트 브라우저가 뜨지만 화면은 제대로 나타나지 않는다. 이 브라우저의 주소창 내용을 복사하여 크로미움(chromium-browser 명령어로 실행)에 붙여넣기를 해야 비로소 리포트를 볼 수 있다.

흥미롭게도 py35 환경에 anvi'o를 설치하면 테스트 스크립트 실행이 완료되면서 Firefox가 뜨고 결과도 전부 제대로 보인다!
다른 문제점
- anvi-display-contigs-stats 스크립트가 없다: py35 environment에 설치하면 된다.
- hmmer가 PATH에 있어야 한다. Anvi'o 버전이 v4 이하이면 hmmer v3.1.b2가 필요하다.
- [python 3.x] anvi-profile –version 명령에서 Anvi'o의 버전이 unknown으로 나온다는 것, 그리고 pangenomics 스크립트(확인된 것은 anvi-compute-ani, anvi-get-enriched-functions-per-pan-group)가 설치되지 않았다는 것. 확인해 보니 이 유틸리티는 anvi'o version 5부터 추가된 것 같다.
python 2.7과 3.x 중 어느것을 기반으로 하느냐에 따라서 db의 버전도 달라진다. Python 3.x 환경에서는 contigs database version이 10으로 바뀌면서 .h5 파일은 쓰이지 않게 된다. 정 문제가 해결되지 않으면 docker image를 써서 실행하자.
[강력 추천!] Docker image 이용하기
Anvi'o의 최신 버전을 쓰고 싶다면 docker image를 적극적으로 활용하라. Interactive output display에 접근하려면 -p 8080:8080을 잊지 말아라.
# docker pull meren/anvio # docker run -p 8080:8080 -it meren/anvio:latest :: anvi`o :: / >>> anvi-self-test --suite mini
여기까지 실행하였으면 다른 터미널 창을 열고 웹브라우저를 구동한 뒤 ip_address:8080을 접속한다. 서버를 종료하려면 Ctrl + C를, host terminal로 돌아오려면 Ctrl + D를 클릭한다.
도커 이미지를 지우고 새로 깨끗하게 설치하려면 docker images를 실행하여 표시되는 이미지의 ID를 찾은 뒤 docker rmi -f <image ID>를 하면 된다.
# docker images REPOSITORY TAG IMAGE ID CREATED SIZE meren/anvio latest b5aab06b73e6 9 days ago 1.452 GB meren/anvio 5 0d6c61689cae 2 weeks ago 1.446 GB ... # docker rmi b5aab06b73e6
HDD의 자료 접근하기
# docker run --rm -v /home/merenbey/my_data:/my_data -it meren/anvio:latest :: anvi'o :: / >>> cd my_data :: anvi'o :: /my_data >>>
Anvi'o로 할 수 있는 일
개요
Anvi'o project page에서 상단의 tutorials를 클릭하면 10 가지가 넘는 각 응용 분야 별의 튜토리얼이 준비되어 있다. 본격적인 실행에 들어가기에 앞서서 먼저 중요한 개념을 몇 가지 정의하고 넘어가자. 직접 de novo assembly를 하여 생성했거나 NCBI에서 다운로드한 genome이 있다면 이를 external genome이라 부른다. 이에 대비되는 internal genome은 anvi;o의 metagenomic analysis 결과로 얻어진 genome bin을 뜻한다. contig 파일은 반드시 .fa로 끝나야 하며, 점(.)은 파일명 안에 단 한번만 나와야 한다. 각 organism마다 contigs database로 전환된 contig는 genomes storage로 전환되어야 비로소 한 덩어리로 취급된다.
COG annotation을 한 다음에는 genomes storage를 다시 만들어서 기능 관련 정보가 채워야 한다.
데이터베이스는 전부 파일로 저장된다. Contigs database에는 각 contig에 부속되는 정보, 즉 k-mer frequency, 20 kb 단위의 soft split, ORF 예측 결과 등이 수록된다(anvi-gen-contig-database). 다른 프로그램으로 생성한 gene call 결과를 이용하는 것 역시 가능하다(–external-gene-calls <TAB-delimited file>; 파일의 포맷은 여기를 참조). 또한 contig 파일의 defline은 최대한 간결하게 고쳐져야 한다. 그러나 anvi-script-FASTA-to-contigs-db 스크립트를 실행하면 이 일이 전부 자동적으로 이루어진다.
정리하자면 FASTA file이 있을 때 바로 다음의 두 과정을 필수적으로 진행해야 한다는 뜻이다.
$ for i in *fa
do
anvi-script-FASTA-to-contigs-db $i
done
(# 필요하다면 anvi-run-ncbi-cogs을 이 단계에서 한다)
$ anvi-gen-genomes-storage -e external-genomes.txt \
-o ALL_GENOMES.h5 (or ALL_GENOMES.db)
external-genomes.txt 파일은 contig database와 실제 이름을 연결하는 정보이다(파일 구조는 여기를 참조). Name은 숫자로 시작해서는 안되며, dash(-)도 허용하지 않는다. 허용되는 것은 ASCII 문자, 숫자, 그리고 밑줄이다. 반면 contig FASTA file의 이름은 확장자만 잘 지키면 된다(.fa). [python 2.7에서만] 실제로 두번째 스크립트(anvi-gen-genomes-storage)를 실행해 보면 genomes storage 파일의 확장자는 .db가 아니라 .h5로 지정하라는 에러 메시지가 나온다.
이전 버전으로 만들어진 contigs database를 업데이트하려면 anvi-migrate-db를 사용한다.
결과물은 결국 interactive interface에서 보는 것이 원칙이다. A tutorial on the anvi'o interactive interface를 참고하라. 그림을 멋지게 꾸미는 소위 'prettification'을 원한다면 꼭 읽어봐야 한다.
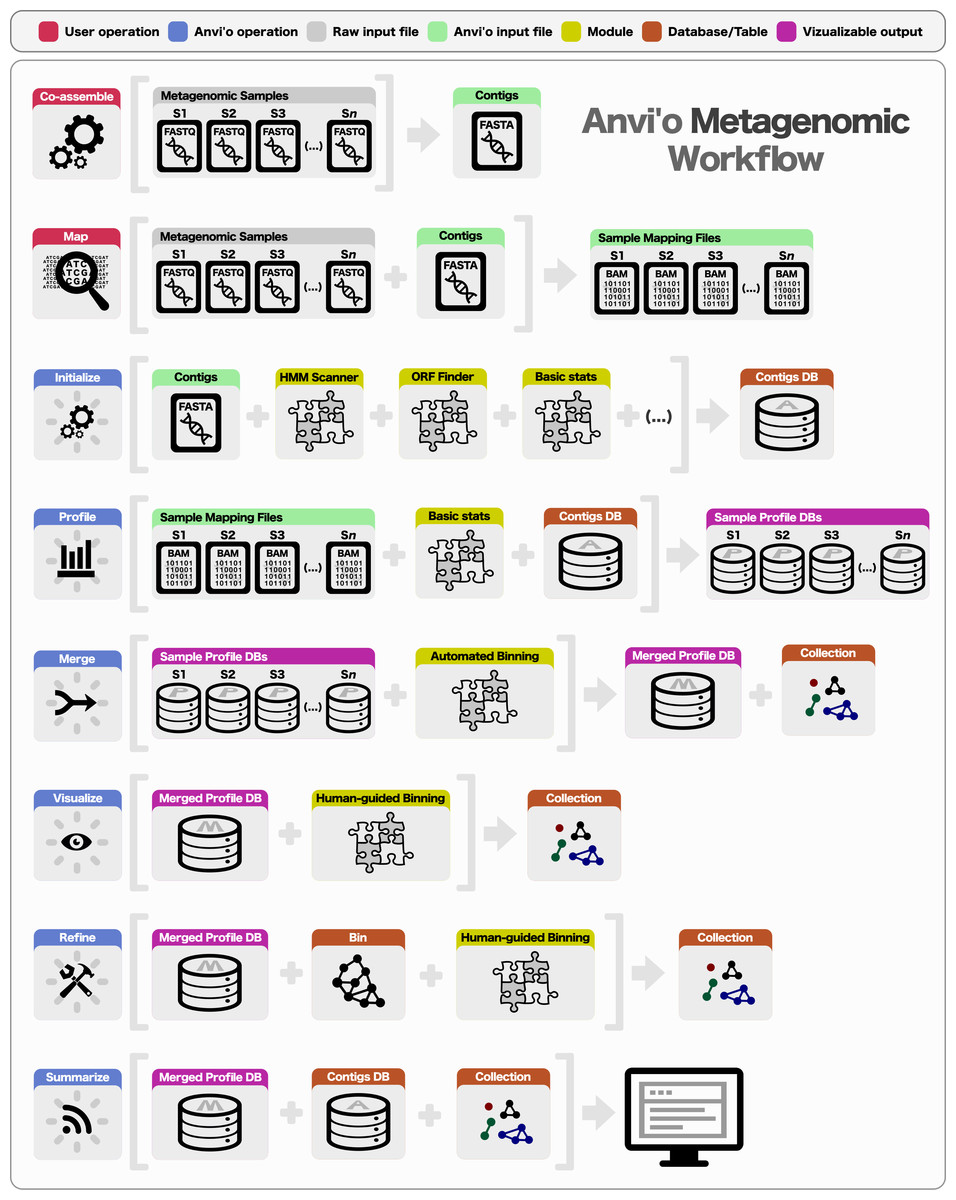
Metagenomics
Pangenomics
An anvi'o workflow for microbial pangenomics An anvi'o workflow for microbial pangenomics (old version)
내가 설치한 anvi'o의 버전을 알고 싶으면 'anvi-profile –version'을 실행한다.
여기에서는 metagenomics workflow 못지않게 상당히 많은 종류의 일을 할 수 있으므로 위 링크에 나오는 tutorial을 자세히 읽기 바란다. 먼저 FASTA file로부터 contigs database를 만들고 이어서 genomes storage(예: MY-GENOMES.db)도 만든 뒤 다음의 스크립트를 실행한다. 이렇게 하면 NCBI BLAST 대신 DIAMOND(default)를 써서 단백질 서열 간 비교를 하는데, 시간은 적게 소요되지만 정확도에서는 불리하다고 한다.
$ anvi-pan-genome -g ALL_GENOMES.db -n PANGENOMES
-n 뒤에 기입하는 것은 PROJECT_NAME으로서 실제로는 이 이름의 디렉토리가 생긴다. 그 하위에 PANGENOMES-PAN.db라는 별도의 DB가 생기는 것이다. 결과를 interactive하게 보려면 다음과 같이 실행한다. 동일 컴퓨터에서 웹브라우저를 띄우는 것보다 다른 컴퓨터(MS-윈도우)에서 크롬으로 접속하는 것이 훨씬 빠르다.
$ anvi-display-pan -p PANGENOMES/PANGENOMES-PAN.db -g ALL_GENOMES.db
Anvi's interactive interface에 대한 상세한 설명은 여기를 참조한다.
Average nucleotide identity(ANI) 분석도 실시할 수 있다.
$ anvi-compute-ani --external-genomes external-genomes.txt \
--output-dir ANI \
--num-threads 6 \
--pan-db PANGENOMES/PANGENOMES-PAN.db
pyani 실행 단계에서 pandas가 없다는 메시지가 나온다. 거듭된 테스트 결과 그 이유를 알아냈다. 도커 이미지 내에서 파이썬 기준 버전은 2.7이다. 그러나 Anvi'o는 3.x 버전에서 돌아가야 하고, pandas도 version 3 기반 위치에 깔려있다. 반면 anvi-compute-ani가 호출하는 average_nucleotide_identity.py(pyani)의 shebang line에는 #/usr/bin/env python이라고 되어있는 것이다. 이를 #!/usr/bin/env python3로 고치면 된다. 그런데 이렇게 하려면 적당한 에디터(예: vim)가 필요하고, docker run –rm 옵션으로 실행을 하면 변경 사항이 저장되지 않음을 유의하라. 그러면 vim을 일부러 설치할 필요는 없다. 근본적인 해결 방법은 변경된 컨테이너를 별도의 이미지로 export하여 재활용하는 것이다.
ANI 분석 결과물의 시각화는 똑같이 anvi-display-pan 명령을 사용한다. 처음에는 ANI 분석 결과가 보이지 않는다. 놀라지 말고 Layout 탭으로 가서 시각화하고 싶은 정보의 체크박스를 클릭한 뒤 Draw를 클릭하라.
External gene call 정보를 이용하기
윗부분 'Anvi'o로 할 수 있는 일'의 개요 항목에 설명하였다.
Layer란?
Working with anvi'o additional data tables
Anvi'o의 interactive display에서는 서열 데이터로부터 anvi'o 작업을 통해 분석된 데이터 외에도 tab-delimited file로 제공되는 외부 데이터도 표시할 수 있다.
Gene cluster 탐색하기
유전자 기능과 관련한 분석하기
Phylogenomics
An anvi'o workflow for phylogenomics
먼저 FASTA file로부터 contigs database를 만든다. genomes storage는 만들 필요가 없다. 다음으로는 marker gene에 대한 HMM hit를 출력하여 concatenated sequence를 만든다. 염기서열이 아니고 아미노산 서열이다.
$ anvi-get-sequences-for-hmm-hits --external-genomes external-genomes.txt \
-o concatenated-proteins.fa \
--hmm-source Campbell_et_al \
--return-best-hit \
--get-aa-sequences \
--concatenate
이렇게 실행하면 Campbell et al.의 139개 마커를 전부 다 사용한다. 이것이 너무 과하다 싶으면 –gene-names maker_1,marker_2,,,로 마커의 수를 제한한다. 다음으로는 phylogenetic tree를 만든다.
$ anvi-gen-phylogenomic-tree -f concatenated-proteins.fa \
-o phylogenomic-tree.txt
Newick format의 tree file이 만들어졌으므로 figtree 등의 소프트웨어로 시각화하거나, 혹은 다음의 방법으로 인터랙티브하게 결과를 띄운다. Manual mode에서는 빈 profile database를 생성하게 된다.
$ anvi-interactive -p phylogenomic-profile.db \
-t phylogenomic-tree.txt \
--title "Phylogenomics Tutorial Example #1" \
--manual
Pangenome 결과 디스플레이에 phylogenomics에서 만든 tree 넣기
COG annoatation
Annotating an anvi'o contigs database with COG
# anvi-setup-ncbi-cogs --num-threads 20
(일반 사용자모드로 전환)
$ for i in *db
do
anvi-run-ncbi-cogs -c $i --num-threads 20
done
COG data directory의 기본 위치는 /usr/local/lib/python3.5/dist-packages/anvio/data/misc/COG이다. 따라서 docker image를 실행하여 설치했다면 exit하는 순간에 지워질 것이다. 따라서 docker commit 명령을 이용하여 설치된 상태의 컨테이너를 별도의 이미지로 저장하는 것이 바람직하다. COG data의 위치는 –cog-data-dir로 변경 가능하다. 그리고 앞에서도 언급했듯이 COG annotation을 한 다음에는 genomes storage를 다시 만들어서 기능 관련 정보가 채워야 한다.
기타 노하우
Docker 안에서 패키지 설치하기
필요하다면 apt-get을 이용하여 필요한 패키지를 설치할 수 있다. 도커 이미지를 처음 적재한 뒤에는 apt-get update를 한번 해 주어야 패키지 설치가 가능하다. 필요한 파이썬 패키지가 있으면 pip3를 사용해야 한다. 기본 인터프리터가 비록 python 2.7이지만 이 이미지에는 pip3가 설치된 상태이고 모든 필요한 라이브러리 역시 이를 기준으로 설치된 상태이다. 도커 컨테이너를 변경하여 재사용하려면 docker commit 명령어를 알아야 한다. 자세한 방법은 [정해영의 블로그] Anvi'o의 사소한 문제점 개선하기 뒷부분을 참조하기 바란다.